بـيـتـا ثـلاسـيـمـيـا (أنيميا البحر المتوسط) Beta Thalassaemia
الثلاسيميا تعني و جود خلل في إنتاج احدى السلاسل الأمينية الضرورية لإنتاج هيموجلوبين البالغين بنسبتة الطبيعية. و يكون الخلل إما في السلسلة ألفا (ألفا ثلاسيميا) أو السلسلة بيتا(بيتا ثلاسيميا) و هي الأكثر إنتشاراً , و يؤدي الخلل إلى نقص في كمية السلسلة المتوفرة لتتحد مع السلاسل من الأنواع الأخرى لإنتاج الهيموجلوبين مما يؤدي إلى ترسبها في كريات الدم الحمراء و بالتالي تكسرها في الطحال أسرع من كريات الدم الحمراء الطبيعية. و الثلاسيميا مرض وراثي يرثه الشخص من الأبوين , و يرث من كل منهما جيناً واحداً لإنتاج السلسلة بيتا, يعني يرث (بصمة وراثية) جين من الأب والآخر من الأم, فإذا ورث جيناً واحداً يكون مصاباً ببيتا ثلاسيميا صُغرى (Beta Thalassemia Trait) , و إذا ورث من الأب و الأم فيكون مصاباً ببيتا ثلاسيميا كُبرى (Beta Thalassemia Major) .
بيتا ثلاسيميا صُغرى
و هي النوع الخفيف و يسمى الشخص "حامل للمرض" و الشخص المصاب تكون حياته طبيعية ونموه طبيعياً, و لكن ربما يشكو من أعراض بسيطة كالدوار أحياناً, أو ضعف عام بسيط, و التحاليل المخبرية تدل على وجود فقر دم بسيط مع زيادة في نسبة هيموجلوبين A2 وأحيانا هيموجلوبين F كذلك.
بيتا ثلاسيميا كُبرى
"مصاب بالمرض" و يؤدي إلى تكسر شديد لكريات الدم الحمراء منذ الصغر و يؤدي إلى تضخم الطحال و الكبد و فقر دم شديد (نقص شديد بكمية الهيموجلوبين في الدم) و يرقان(بو صفار) و زيادة كمية الحديد في الجسم و ترسبه في الأنسجة مما يؤدي إلى تلفها.هيموجلوبين البالغين لا يوجد في الدم أو موجوداً بنسبة ضئيلة جداً و السائد هو الهيموجلوبين الجنيني (هيموجلوبين Haemoglobin F)و يحتاج المريض إلى نقل دم طيلة حياته.
مُـضـاعـفـات الـمـرض :
1- تضخم نخاع العظم في محاولته إنتاج كريات دم حمراء لتعويض ما يتكسر منها مما يؤدي إلى هشاشة العظام و قابليتها للكسر بسرعة, و كذلك تغير ملامح وجه المصاب ليبدو شبيها بالمنغوليين.
2- تضخم الطحال مما يزيد من حدة تكسر كريات الدم الحمراء فتزيد الحالة سوء و كذلك الطحال المتضخم يكون أكثر عرضة للتمزق و النزيف إذا ما تعرض المريض لضربة على بطنه أيا كان سببها.
3- زيادة الحديد في الجسم (Haemochromatosis) من عملية نقل الدم و تكسر كريات الدم الحمراء و ترسبه في الأنسجة مما يؤدي إلى تلفها و هبوط عملها و المرض الذي ينتج عن ذلك يعتمد على نوع العضو المصاب:
- الغدة الدرقية و الجار درقية مما يؤدي إلى هبوط في عملهما (Hypothroidism – Hypoparathyroidism).
- فشل الكبد و التأثير على وظائفها ("Hepatic Failure "Cirrhosis).
- فشل القلب (Heart Failure).
- البنكرياس و يؤدي ذلك إلى تلفها ثم عدم مقدرتها على إفراز الأنسولين مما يؤدي إلى الإصابة بمرض السكري Diabetes Mellitus, و يترسب الحديد كذلك في الجلد فيكسبه لوناً برونزياً, لذا يطلق اسم مرض السكري البرونزي (Bronze Diabetes) على هذا النوع.
- أمراض تنتقل نتيجة نقل الدم مثل التهاب الكبد الفيروسي (Viral Hepatitis) و كذلك مرض الإيدز (AIDS).
التشخيص: يكون عن طريق تحليل أنواع الهيموجلوبين الموجودة في الدم (Haemoglobin Electrophoresis).
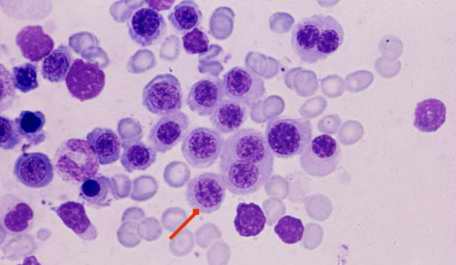
صورة مجهرية لدم مصاب بثلاسيميا كبرى.
لاحظ وجود خلايا سليفات الحمراء (ذوات النواة) و التي لا تكون موجودة في الدم الطبيعي
العلاج:
1- نقل الدم تقريبا شهريا للمحافظة على مستوى معقول للهيموجلوبين في الدم.
2- التخلص من الحديد الزائد في الجسم بإعطاء أدوية تتحد معه ليطرح خارج الجسم مثل ديسفيرال (Desferrioxamine) .
3- أخذ أقراص الفوليت (Folic acid) لتجنب النقص فيه و ذلك لزيادة طلب الجسم نتيجة لتكسر كريات الدم الحمراء و أهميتة في إنتاجها في الجسم.
4- زراعة نخاع العظم (Bone Marrow Transplant) و يجب توفر متبرع ملائم مطابق من حيث التركيبة الوراثية.
5- العلاج بالجينات , أي تعديل التركيبة الجينية(الوراثية) للمريض (Gene Therapy) في المستقبل حيث أن التركيز على العلاج الجيني هو محور الأبحاث في كل الأمراض.
الناحية الوراثية:
و هي الناحية المهمة, و يجب على المصاب فهمها و إدراكها, و أكثر الأمراض الوراثية تنتج عن تزاوج الأقارب, حيث تكون نسبة اتحاد الجينات المكبوتة (Recessive Genes) عالية.
سوف نرمز للجين السليم بß و الجين الغير سليم ب ß o فيكون رمز بيتا ثلاسيميا صغرى
(ßß o ) و ثلاسيميا كبرى (ßo ßo ) و الشخص السليم (ß ß ) :
1- فإذا تم الزواج بين شخص سليم (ß ß ) و آخر مصاب ببيتا ثلاسيميا صغرى (ß ß o ) حامل للمرض تكون الاحتمالات كما يلي:
|
ßo |
ß |
|
|
ßßo |
ßß |
ß |
|
ßßo |
ßß |
ß |
أي أن إحتمال إصابة الجنين في الحمل ببيتا ثلاسيميا صغرى تكون 50% (حامل للمرض).
2- إذا تم الزواج بين شخص حامل للمرض (ßßo) من آخر مصاب بنفس الحالة (ßßo ) حامل للمرض تكون الاحتمالات كما يلي:
|
ßo |
ß |
|
|
ßßo |
ßß |
ß |
|
ßoßo |
ßßo |
ßo |
أي أن إحتمال إصابة الجنين في الحمل ببيتا ثلاسيميا صغرى تكون 50% (حامل للمرض), و 25% بببيتا ثلاسيميا كبرى و 25% طبيعي.
3- إذا تم الزواج بين شخص مصاب بالمرض (ßo ßo) من شخص سليم (ßß ) فان جميع الأولاد يكونون حاملين للمرض (بيتا ثلاسيميا صغرى):
|
ßo |
ßo |
|
|
ßßo |
ßßo |
ß |
|
ßßo |
ßßo |
ß |
4- إذا تم الزواج بين شخص مصاب بالمرض (ßoßo) من شخص حامل للمرض (ßßo ) فان الاحتمالات تكون كما يلي:
|
ßo |
ßo |
|
|
ßßo |
ßßo |
ß |
|
ßo ßo |
ßoßo |
ßo |
أي أن إحتمال إصابة الجنين في الحمل بببيتا ثلاسيميا كبرى تكون50%و 50% بببيتا ثلاسيميا صغرى.
5- وإذا تزوج شخص مصاب من آخر مصاب كذاك فإن جميع الأولاد يصابون ببيتا ثلاسيميا كبرى.
و هناك حالات تنتج من إتحاد الجينات (البصمات الوراثية) المختلفة بعضها ببعض الموروثة من الأبوين. فإذا كان الأب مثلا حاملا للثلاسيميا, و الأم حاملة للمنجليه ينتج عن ذلك مرض بيتا ثلاسيميا/منجليه (ٍSickle/Thalassemia) , كذلك يمكن أن يكون نفس الشخص مصاباً بنفص أنزيم G6PD لأنه يورث عن طريق آخر و هو الكروموسوم الجنسي.
مما سبق يتضح أهمية التأكد من خلو الطرف الثاني في الزواج من بيتا ثلاسيميا صغرى إذا كان الطرف الأول حاملا لها, و ذلك لتجنب إنجاب أطفالا مصابين ببيتا ثلاسيميا كبرى, مما يؤدي إلى معاناة الطفل والأبوين.
و يستحسن أن يتأكد أي شخص يريد الزواج من خلوه هو نفسه من هذه الأمراض و خلو الطرف الآخر منها, و بخاصة مع وجود تاريخ عائلي للمرض حتى لو كانا لا يشتكون من أية أعراض خلال حياتهما.
د.خليل رضا اليوسفي
استشاري طب العائلة - الكويت